La FDA n’exige pas toujours des études sur des humains pour prouver que deux médicaments sont équivalents. Dans certains cas, elle accepte des données issues de tests en laboratoire. C’est ce qu’on appelle une dérogation à l’équivalence biologique, ou biowaiver. Pourquoi ? Parce que pour certains médicaments, les tests en éprouvette sont plus fiables, plus rapides et moins coûteux que les essais sur des patients.
Qu’est-ce qu’une dérogation à l’équivalence biologique ?
Une dérogation à l’équivalence biologique permet à un fabricant de ne pas réaliser d’étude in vivo - c’est-à-dire une étude sur des volontaires humains - pour prouver que son médicament générique se comporte comme le médicament de référence. Au lieu de cela, il peut se fier à des données de dissolution en laboratoire. Cela ne signifie pas que la FDA baisse les normes. Au contraire : elle exige que les tests in vitro soient les plus précis, sensibles et reproductibles possibles, comme le stipule le règlement 21 CFR 320.24(a).
Avant cette dérogation, chaque nouveau générique devait passer par une étude in vivo coûteuse : des volontaires prenaient le médicament, on mesurait leur taux sanguin pendant des heures, et on comparait les courbes d’absorption. Ces études coûtent entre 250 000 et 500 000 dollars, et prennent 6 à 12 mois. Avec une dérogation, ce processus disparaît - si les critères sont remplis.
Quels médicaments sont concernés ?
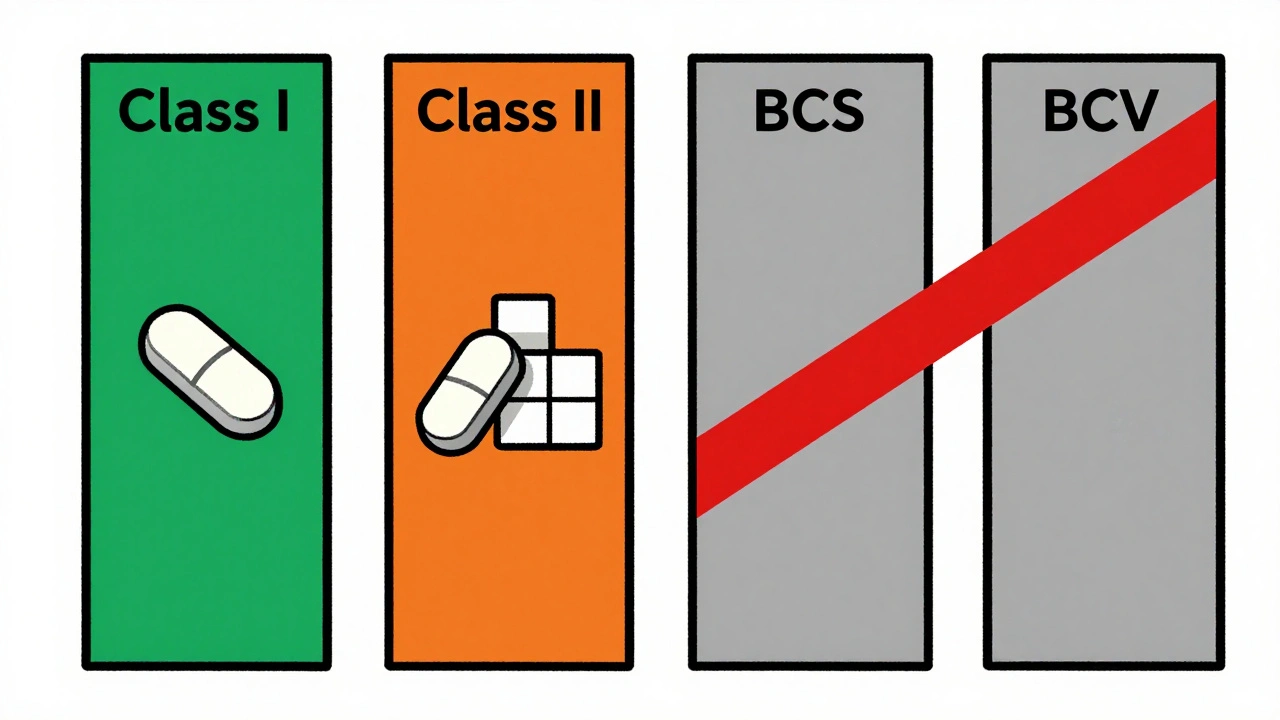
Les dérogations ne s’appliquent qu’à un type très précis de médicaments : les formes orales solides à libération immédiate. Pas les comprimés à libération prolongée, pas les gélules à effet ciblé, pas les solutions injectables. Et encore, seulement si le principe actif entre dans deux catégories du Système de Classification Biopharmaceutique (BCS).
Le BCS classe les médicaments selon deux critères : leur solubilité dans les fluides corporels et leur perméabilité à travers la paroi intestinale. Seuls deux groupes peuvent prétendre à une dérogation :
- Classe I : hautement solubles et hautement perméables. Exemples : l’ibuprofène, la metformine, le propranolol. Pour ces médicaments, la dissolution est le seul frein à l’absorption. Si la forme générique se dissout aussi vite que le médicament de référence, elle sera absorbée de la même manière.
- Classe III : hautement solubles mais faiblement perméables. Exemples : l’acide aminocaproïque, la cimétidine. Ici, la perméabilité est le problème. La FDA exige alors des conditions encore plus strictes : mêmes excipients, mêmes proportions, et preuve que l’absorption ne dépend pas de l’endroit dans l’intestin.
Les classes II et IV - faiblement solubles - ne sont pas concernées. Pour ces médicaments, la dissolution est trop imprévisible pour être remplacée par un test en laboratoire.
Quels tests en laboratoire sont exigés ?
Le cœur de la dérogation est un test de dissolution. Il ne suffit pas de dire : "ça se dissout bien". Il faut montrer que la forme générique se dissout exactement comme le médicament de référence, dans des conditions qui imitent l’intestin humain.
La FDA exige :
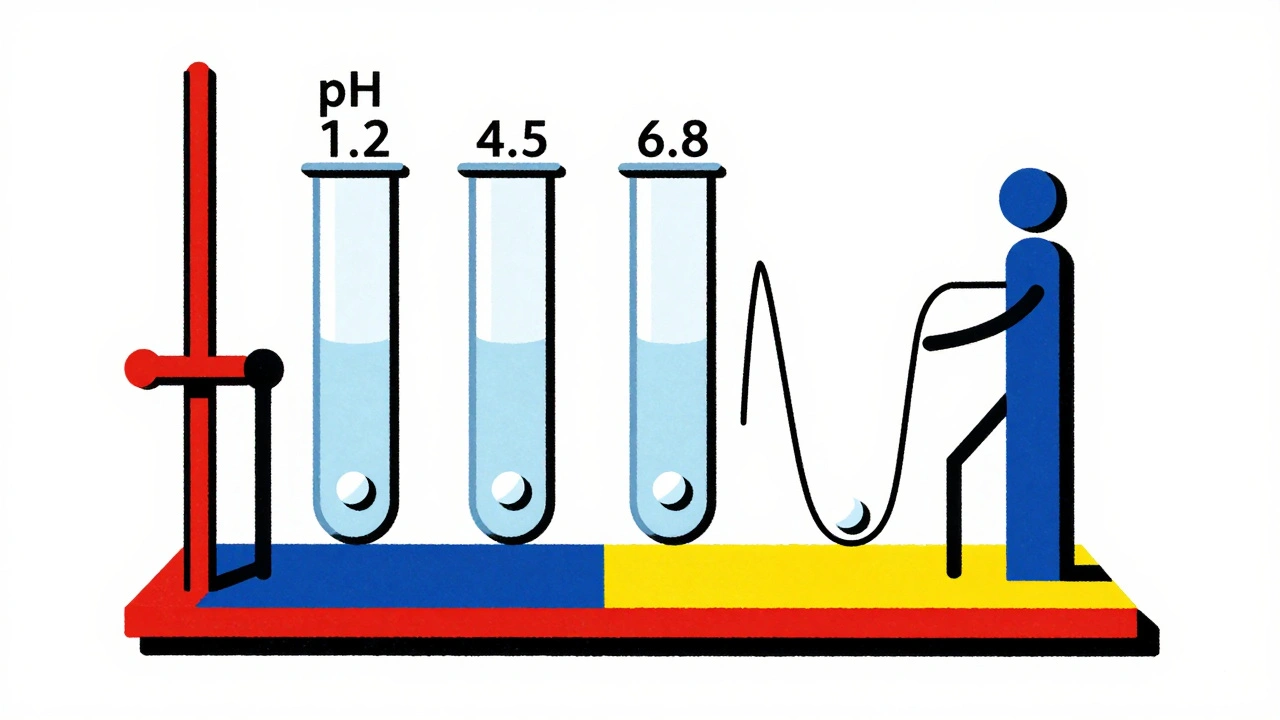
- Des tests dans trois tampons : pH 1.2 (estomac), pH 4.5 (duodénum), et pH 6.8 (intestin grêle).
- Un minimum de 12 unités par formulation (comprimés ou gélules).
- Des prélèvements à 10, 15, 20, 30, 45 et 60 minutes.
- Un facteur de similarité f2 ≥ 50 entre les courbes de dissolution du générique et du produit de référence.
Le facteur f2 est une mesure statistique. Plus il est proche de 100, plus les courbes sont identiques. Un f2 ≥ 50 signifie que les deux produits se dissolvent de manière suffisamment similaire pour être considérés équivalents. Si le f2 est à 48, la demande est rejetée - même si le médicament est chimiquement identique.
Quand les dérogations sont-elles rejetées ?
La FDA approuve environ 78 % des demandes de dérogation quand elles sont bien préparées. Mais 35 % des rejets sont dus à un seul problème : le test de dissolution n’est pas suffisamment discriminant.
Qu’est-ce que ça veut dire ? Cela signifie que le test ne parvient pas à détecter les différences subtiles entre les formulations. Par exemple, si deux comprimés se dissolvent à 85 % en 30 minutes dans un tampon pH 6.8, mais que l’un contient un excipient qui ralentit l’absorption dans l’intestin, le test doit le révéler. Si le test est trop doux, il ne le voit pas. Et la FDA ne peut pas accepter un test qui ne voit pas les différences.
Autre raison fréquente de rejet : les excipients ne sont pas identiques pour les médicaments de classe III. Même un petit changement dans le type de cellulose ou de lubrifiant peut affecter la perméabilité. Et la FDA ne laisse pas passer ça.
Quels sont les avantages réels ?
Les entreprises de génériques utilisent les dérogations pour gagner du temps et de l’argent. Entre 2012 et 2016, 15 % des demandes d’ANDA (demandes de génériques) incluaient une demande de dérogation. Aujourd’hui, ce chiffre est passé à 18 %. Et les résultats sont tangibles :
- Une dérogation permet d’économiser entre 400 000 et 600 000 dollars par produit.
- Elle accélère l’approbation de 8 à 10 mois.
- En 2022, l’usage des dérogations a permis d’entrer plus tôt sur le marché pour 1,2 milliard de dollars de génériques.
Des sociétés comme Teva et Mylan intègrent les dérogations dans 25 à 30 % de leurs projets. Pour les petites entreprises, c’est plus difficile : elles manquent souvent de scientifiques expérimentés pour développer les tests de dissolution. Le processus prend 2 à 3 mois de développement, puis 1 à 2 mois de validation. Sans équipe spécialisée, la demande échoue.

Quels sont les limites et les controverses ?
Les dérogations ne s’appliquent pas aux médicaments à index thérapeutique étroit - comme la warfarine, le lithium ou la phénytoïne - sauf pour quelques cas spécifiques. Pourquoi ? Parce qu’un léger écart d’absorption peut causer une overdose ou un échec thérapeutique. La FDA ne prend pas ce risque.
Les experts s’interrogent aussi sur la rigidité du BCS. Certains, comme la Dr. Jennifer Dressman, estiment que la classification exclut trop de médicaments de la classe II, qui pourraient pourtant être évalués par des tests in vitro avancés. Des essais pilotes sont en cours pour étendre les dérogations à certains produits complexes, mais pour l’instant, 85 % des génériques complexes restent exclus.
Un autre problème : l’application inégale des critères. Selon une enquête de PhRMA en 2022, 42 % des entreprises ont rencontré des différences entre les équipes de révision de la FDA. Une demande approuvée à un endroit est rejetée à un autre, sans raison claire. Cela crée de l’incertitude pour les fabricants.
Comment augmenter ses chances d’obtenir une dérogation ?
Si vous êtes un fabricant, voici comment faire pour maximiser vos chances :
- Commencez par classer votre principe actif selon le BCS. Utilisez les données publiées par la FDA ou des bases comme BCS Gateway.
- Si vous êtes en classe I ou III, développez un test de dissolution avec au moins trois tampons et un minimum de 12 unités.
- Validez la méthode : montrez qu’elle détecte les différences entre formulations. Testez des lots avec des variations contrôlées.
- Comparez vos courbes de dissolution avec celles du médicament de référence. Calculez le f2. Il doit être ≥ 50.
- Ne négligez pas les excipients. Pour la classe III, ils doivent être identiques en type et en quantité.
- Demandez une réunion préalable via le programme Pre-ANDA. Les dossiers avec réunion ont 22 % plus de chances d’être approuvés.
La FDA encourage aussi les entreprises à consulter les guides techniques publiés par l’ICH (M9) et à s’appuyer sur les données publiées dans les revues comme le AAPS Journal, qui montre une concordance de plus de 95 % entre les dérogations et les études in vivo pour les classes I.
Quel avenir pour les dérogations ?
La FDA a fixé un objectif : augmenter de 25 % les dérogations scientifiquement justifiées d’ici 2027. Des projets sont en cours pour étendre les dérogations aux classes II avec des modèles de corrélation in vitro-in vivo (IVIVC) plus avancés. Des essais pilotes testent aussi les dérogations pour certains médicaments à index thérapeutique étroit.
Le financement est là : 15 millions de dollars par an sont alloués au programme GDUFA pour développer de nouveaux tests de dissolution. La science avance. Les méthodes deviennent plus sensibles. Les algorithmes de prédiction s’améliorent.
Le message est clair : la FDA ne veut pas de tests inutiles. Elle veut des preuves fiables. Et si un test en laboratoire peut faire aussi bien - voire mieux - qu’une étude sur des humains, elle l’acceptera. Pourvu que la science soit solide.
Quelle est la différence entre une étude in vivo et une dérogation à l’équivalence biologique ?
Une étude in vivo mesure la concentration d’un médicament dans le sang de volontaires humains pour prouver qu’il est absorbé de la même manière que le médicament de référence. Une dérogation à l’équivalence biologique (biowaiver) permet de remplacer cette étude par des tests de dissolution en laboratoire, à condition que le principe actif soit classé BCS I ou III et que les courbes de dissolution soient identiques. La dérogation évite les essais sur des humains, mais exige des données in vitro plus rigoureuses.
Pourquoi la FDA accepte-t-elle les dérogations pour les BCS I mais pas pour les BCS II ?
Les médicaments BCS I sont hautement solubles et hautement perméables. Leur absorption est limitée uniquement par la vitesse à laquelle ils se dissolvent. Si deux comprimés se dissolvent de la même manière, ils seront absorbés de la même manière. Les BCS II, en revanche, sont mal solubles. Leur absorption dépend de facteurs complexes comme la motilité intestinale, la présence de graisses, ou les variations de pH. Ces facteurs ne peuvent pas être simulés de manière fiable en laboratoire. Donc, la FDA exige encore des études in vivo pour ces médicaments.
Les dérogations sont-elles acceptées dans l’Union européenne ?
Oui. L’Agence européenne des médicaments (EMA) suit les mêmes principes que la FDA, basés sur le Système de Classification Biopharmaceutique (BCS) et les lignes directrices de l’ICH M9. Les critères pour les dérogations sont très similaires : solubilité élevée, perméabilité élevée, et profils de dissolution comparables. Les entreprises peuvent utiliser les mêmes données pour des demandes aux États-Unis et en Europe.
Combien de temps faut-il pour développer un test de dissolution valide ?
Pour une équipe expérimentée, le développement d’un test de dissolution approprié prend entre 2 et 3 mois. Cela inclut la sélection des tampons, la validation de la méthode, et la démonstration qu’elle est discriminante. Ensuite, il faut 1 à 2 mois supplémentaires pour réaliser les comparaisons avec le produit de référence et préparer le dossier. Les entreprises sans expertise en biopharmacie mettent souvent 6 mois ou plus.
Les dérogations sont-elles fiables ?
Oui, pour les BCS I. Une étude publiée dans le AAPS Journal en 2020 a montré que les dérogations ont une concordance de plus de 95 % avec les résultats des études in vivo. Cela signifie que dans 19 cas sur 20, un générique approuvé par dérogation se comporte exactement comme le médicament original. Pour les BCS III, la concordance est plus faible, mais toujours supérieure à 85 % si les excipients sont identiques. La fiabilité dépend donc de la rigueur scientifique du test.


12 Commentaires
Marc LaCien
C’est fou comment la science peut simplifier la vie sans sacrifier la sécurité 🚀
Gerard Van der Beek
jai lu ca 3 fois et j’ai encore pas compris pourquoi les classe II sont exclues… c’est pas un peu arbitraire ?
Yacine BOUHOUN ALI
Ah, le BCS… cette classification qui fait rêver les pharmacologues et paniquer les développeurs de génériques. Ce n’est pas juste une règle, c’est une philosophie de la bioéquivalence. Et pourtant, combien de fois avons-nous vu des formulations identiques en laboratoire mais comportementalement différentes chez l’humain ? La science est belle… mais l’organisme, lui, n’aime pas les simplifications.
Blanche Nicolas
Je suis tellement fière de voir à quel point la science française et européenne contribue à ça ! L’EMA et la FDA en parfaite harmonie… c’est rare et précieux 💖
Sylvie Bouchard
Moi j’adore quand la régulation devient plus intelligente. Au lieu de tout tester sur des humains, on utilise la science pour éviter les tests inutiles. C’est plus éthique, plus rapide, et ça fait baisser les prix. Pourquoi on n’applique pas ça à tout ?
Philo Sophie
Je me demande si les petites entreprises ont vraiment accès à ces données. Ou si c’est juste un jeu pour les gros acteurs avec des laboratoires internes.
Manon Renard
La fiabilité à 95 % pour les BCS I est impressionnante… mais je me demande si ce chiffre ne masque pas une sous-estimation des variations individuelles. Ce n’est pas parce que 19 sur 20 marchent qu’on peut ignorer le 1/20.
James Harris
f2 >= 50 ? C’est le seuil magique. Si t’as 49, t’es bon pour 18 mois de retards. La FDA, c’est comme un prof de maths : il veut la bonne réponse, pas la jolie.
Jacque Johnson
Je trouve ça incroyable qu’on puisse éviter des essais sur des humains pour des médicaments aussi courants. C’est une avancée majeure pour l’accès aux soins. Bravo à ceux qui ont travaillé là-dessus !
Angelique Manglallan
Oui, tout ça c’est très joli… mais dis-moi, combien de génériques approuvés par dérogation ont fini en procès parce qu’un patient a eu une réaction inattendue ? La science peut être précise… mais le corps humain, lui, n’aime pas les chiffres. Il veut des garanties. Et les garanties, ça se teste avec du sang, pas avec des tampons.
Micky Dumo
Selon les lignes directrices ICH M9, la validation du test de dissolution doit inclure la démonstration de la spécificité, de la linéarité, de la précision et de la robustesse. Toute demande de biowaiver doit impérativement intégrer ces paramètres dans le cadre de la validation méthodologique. La non-conformité à ces critères constitue un rejet systématique.
Philippe Lagrange
tu crois que la FDA va un jour accepter les dérogations pour les médicaments en spray nasal ? j’ai lu un truc sur un papier de 2023 qui disait que c’était possible avec des modèles de simulation… mais bon, j’ai peut-être mal lu 😅