
Les médicaments biologiques, comme Humira ou Enbrel, coûtent des dizaines de milliers de dollars par an. Pourtant, leur version moins chère - le biosimilaire - ne peut pas arriver sur le marché avant une longue période d’exclusivité. Pourquoi ? Parce que la loi américaine, le Biologics Price Competition and Innovation Act (BPCIA) de 2009, a créé un système complexe qui protège les fabricants originaux pendant 12 ans, même après l’expiration des brevets principaux.
Les deux couches de protection : données et marché
La loi BPCIA ne se contente pas de protéger les brevets. Elle ajoute deux couches de protection distinctes. La première, appelée exclusivité des données, interdit aux fabricants de biosimilaires de soumettre leur demande d’autorisation à la FDA pendant les 4 premières années après la mise sur le marché du produit original. La seconde, exclusivité du marché, empêche la FDA d’approuver un biosimilaire pendant 12 ans, même si la demande a été déposée.
Cela signifie que même si un brevet expire après 7 ans, le biosimilaire ne peut toujours pas être vendu avant la fin des 12 ans. Ce système n’existe pas pour les médicaments génériques classiques. Un générique d’un médicament chimique peut être approuvé dès l’expiration du brevet, souvent en moins de deux ans. Les biologiques, eux, sont des molécules vivantes, complexes, produites dans des cellules vivantes. Leur fabrication est difficile à reproduire exactement - c’est pourquoi la FDA exige des preuves supplémentaires de similitude.
Le « patent dance » : une bataille juridique cachée
Après que le fabricant de biosimilaire a déposé sa demande, une procédure appelée le « patent dance » commence. C’est un échange obligatoire d’informations entre le fabricant original et le fabricant du biosimilaire. Le premier doit fournir des détails sur ses brevets, et le second doit répondre point par point, en disant quels brevets il conteste et pourquoi.
Cette procédure est censée favoriser les négociations. En réalité, elle est souvent utilisée pour retarder les biosimilaires. Les grandes entreprises comme AbbVie ont accumulé des centaines de brevets pour un seul médicament - 166 pour Humira, selon l’ONG I-MAK. Chaque brevet peut être utilisé pour déclencher un procès. Les litiges peuvent durer des années. En 2023, Humira n’avait toujours pas de biosimilaire aux États-Unis, alors que des versions génériques étaient disponibles en Europe depuis 2018.
Coûts pour les patients et le système de santé
Le retard d’entrée des biosimilaires a un coût humain. Aux États-Unis, le prix de Humira a augmenté de 470 % entre 2012 et 2022. En Europe, où les biosimilaires sont arrivés plus tôt, les prix sont restés stables. Des études montrent que 63 % des pharmaciens ont des patients qui abandonnent leur traitement parce qu’ils ne peuvent pas le payer.
Le Bureau du Budget du Congrès estime que si les barrières à l’entrée des biosimilaires étaient réduites, le système de santé américain pourrait économiser 158 milliards de dollars d’ici 2034. À l’heure actuelle, avec les retards actuels, les économies prévues ne dépassent pas 71 milliards. Ce gap de 87 milliards représente des traitements non accessibles, des douleurs non soulagées, des hospitalisations évitables.

Les biosimilaires sont plus chers et plus longs à développer
Contrairement aux génériques, qui coûtent entre 1 et 2 millions de dollars à développer, un biosimilaire peut coûter plus de 100 millions et prendre entre 5 et 9 ans. Pour les biologiques les plus complexes - comme les anticorps conjugués ou les thérapies cellulaires - les coûts dépassent 250 millions. Cela explique pourquoi peu d’entreprises osent entrer sur ce marché.
En plus du coût, la réglementation est lourde. La FDA exige des études analytiques, des études pharmacocinétiques, et parfois des essais cliniques comparatifs pour prouver qu’il n’y a « aucune différence cliniquement significative » entre le produit original et le biosimilaire. Cela demande des laboratoires spécialisés, des équipements coûteux, et des experts en biologie moléculaire.
Le « vide des biosimilaires » : 118 biologiques en péril, seulement 12 en développement
Entre 2025 et 2034, 118 biologiques perdront leur protection. Leur marché total représente 234 milliards de dollars. Pourtant, seulement 12 d’entre eux ont un biosimilaire en développement. Pourquoi ?
- 32 % ont un faible potentiel de vente - les entreprises préfèrent investir dans des médicaments plus rentables.
- 47 % sont bloqués par des brevets complexes ou des litiges en cours.
- 28 % sont utilisés en oncologie, où les protocoles sont rigides et les médecins hésitent à changer.
- 64 % sont des médicaments pour maladies rares (orphan drugs). Pour ces traitements, les marchés sont petits, les risques élevés, et les investisseurs se retirent.
Seul un biosimilaire pour eculizumab, un traitement pour une maladie rare, est en cours de développement. Sur les 88 % des biologiques orphelins qui vont bientôt perdre leur protection, aucun n’a de biosimilaire en vue. Ce n’est pas un oubli. C’est un échec systémique.
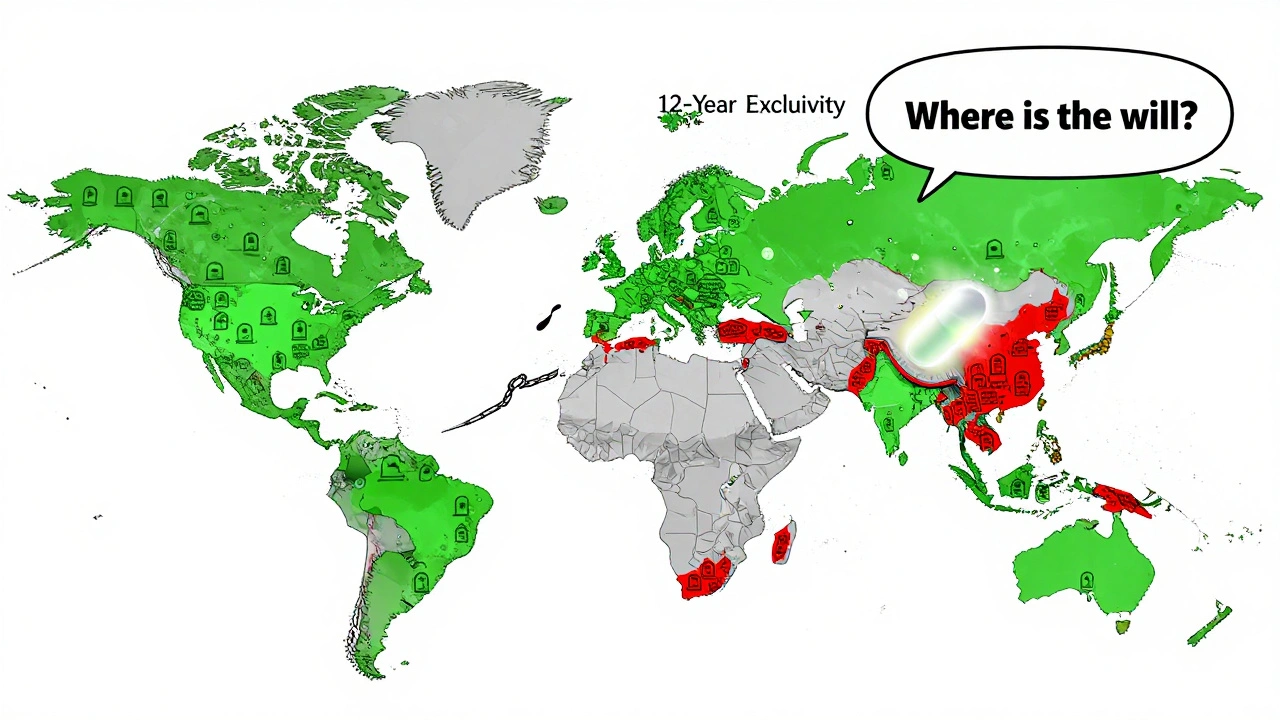
Les États-Unis sont en retard sur l’Europe
En Europe, les biosimilaires représentent maintenant 72 % du marché pour les biologiques avec une version générique disponible. Le processus y est plus simple : 10 ans d’exclusivité des données, puis 1 an d’exclusivité du marché. Pas de « patent dance » obligatoire. Pas de 12 ans d’attente. Les médecins et les patients y sont plus habitués aux biosimilaires. Les prix ont chuté de 40 à 80 % dans les premières années.
En Amérique, seulement 38 biosimilaires ont été approuvés depuis 2015. En Europe, ce chiffre est de 88. La différence n’est pas technologique. C’est une question de politique. Les États-Unis ont choisi de privilégier les profits des entreprises sur l’accès des patients.
Que peut-on faire ?
La FDA a publié un plan d’action en 2022 pour améliorer la transparence, simplifier les processus et encourager la concurrence. Mais jusqu’à présent, les changements sont minimes. Des projets de loi comme le « Biosimilars User Fee Act of 2022 » sont restés bloqués au Congrès.
Les experts proposent plusieurs solutions : réduire l’exclusivité des données à 8 ans (comme au Japon), interdire les « patent thickets » pour les médicaments orphelins, ou créer des incitations financières pour les biosimilaires dans les maladies rares. Sans changement, des millions de patients continueront à payer des prix exorbitants pour des traitements qui pourraient coûter 10 fois moins.
Le temps de l’attente est fini. Les brevets vont expirer. Les biosimilaires sont prêts. Ce qui manque, c’est la volonté politique.
Combien de temps dure l’exclusivité des données pour un biologique aux États-Unis ?
Aux États-Unis, l’exclusivité des données pour un biologique dure 4 ans. Pendant cette période, les fabricants de biosimilaires ne peuvent pas soumettre leur demande d’autorisation à la FDA. Cette règle est prévue par le BPCIA de 2009 et s’applique à tous les produits biologiques approuvés après cette date.
Pourquoi les biosimilaires ne sont-ils pas aussi courants aux États-Unis qu’en Europe ?
En Europe, les règles sont plus simples : 10 ans d’exclusivité des données, puis 1 an d’exclusivité du marché. En plus, il n’y a pas de procédure obligatoire de « patent dance ». Les médecins et les patients y sont plus habitués aux biosimilaires, et les prix baissent rapidement. Aux États-Unis, la combinaison d’une exclusivité de 12 ans, de litiges prolongés et d’un manque de confiance des prescripteurs freine leur adoption.
Qu’est-ce qu’un « patent thicket » et comment cela bloque-t-il les biosimilaires ?
Un « patent thicket » est un ensemble de centaines de brevets secondaires déposés autour d’un même médicament - par exemple, des brevets sur des méthodes de fabrication, des formes galéniques, ou des utilisations spécifiques. Ces brevets n’ont pas toujours un lien direct avec l’efficacité du médicament, mais ils permettent aux entreprises de déclencher des procès contre les biosimilaires. AbbVie a déposé 166 brevets pour Humira, ce qui a retardé l’entrée des biosimilaires aux États-Unis jusqu’en 2023.
Les biosimilaires sont-ils aussi sûrs que les biologiques d’origine ?
Oui. La FDA exige que les biosimilaires soient « hautement similaires » aux produits d’origine, avec « aucune différence cliniquement significative » en termes de sécurité, de pureté et d’efficacité. Des milliers de patients ont déjà reçu des biosimilaires aux États-Unis et en Europe, et les données de sécurité sont comparables à celles des biologiques d’origine. Leur efficacité est prouvée par des études cliniques rigoureuses.
Pourquoi les biosimilaires pour les maladies rares sont-ils si rares ?
Les maladies rares concernent peu de patients, donc le marché est petit. Développer un biosimilaire coûte plus de 100 millions de dollars. Pour une maladie rare, les revenus potentiels ne justifient pas cet investissement. De plus, les réglementations sont plus complexes, et les essais cliniques sont plus difficiles à mener. Résultat : 88 % des biologiques pour maladies rares qui vont perdre leur protection n’ont aucun biosimilaire en développement.

9 Commentaires
Anabelle Ahteck
Les biosimilaires c’est bien mais qui va payer les labos qui les font ? Les big pharma ont des milliards à protéger et les petits ont pas les moyens de se battre contre ça
Yves Merlet
Je suis médecin en région parisienne, et je peux vous dire que les biosimilaires, c’est une révolution pour les patients ! Ils sont aussi sûrs, souvent plus abordables, et ça change la vie de gens qui devaient choisir entre manger ou prendre leur traitement. La FDA les valide à la perfection, et les Européens ont déjà prouvé que ça marche. Il faut arrêter de croire que plus cher = meilleur. C’est une idée reçue dangereuse !
Jonas Jatsch
Je trouve ça incroyable qu’on puisse encore débattre de ça en 2025. On parle de vies humaines, pas de chiffres de Bourse. Les 12 ans d’exclusivité sont une escroquerie légale, et le « patent dance » est un stratagème juridique pour ralentir la concurrence déloyale. Les entreprises comme AbbVie n’ont pas inventé Humira pour soigner les gens - elles l’ont inventé pour faire des profits. Et maintenant, elles utilisent des centaines de brevets bidons pour bloquer des traitements qui pourraient sauver des millions. C’est du capitalisme sauvage, et ça ne peut plus durer. La France et l’Europe doivent imposer des sanctions, pas des recommandations douces. Le système est corrompu, et les patients paient le prix fort.
Kate Orson
Et si c’était juste une manipulation de l’OMS et de la FDA pour faire entrer des médicaments chinois de mauvaise qualité ? 😏 Vous avez vu comme ils trichent sur les normes ? Les biosimilaires, c’est le début du contrôle mondial des médicaments. Un jour, ils vont nous forcer à prendre des injections « équivalentes » faites dans un laboratoire de Shenzhen… avec des QR codes pour suivre nos anticorps. 🤖💊 #BigPharmaIsWatching
Nicole Gamberale
Oh mon Dieu, encore une fois les Américains qui veulent tout garder pour eux pendant que les Européens font preuve de bon sens. 😒 C’est pas une question de science, c’est une question de moralité. Vous avez vu le prix de Humira ? C’est du vol à main armée. Et vous, les gars qui défendez les big pharma, vous avez déjà eu un enfant avec une maladie auto-immune ? Non ? Alors fermez votre gueule. 🤬 Les biosimilaires, c’est la justice sociale en pharmacie. Point.
Alexis Butler
Vous oubliez que la complexité des biologiques justifie pleinement les coûts et les délais. Un générique, c’est une molécule simple, un biosimilaire, c’est une œuvre d’art biologique. La FDA exige des études de bioéquivalence à l’échelle moléculaire, des essais sur des populations de milliers de patients, des analyses de glycosylation… Ce n’est pas comme faire un paracetamol. Si vous pensez que c’est facile, vous n’avez jamais lu un protocole d’essai clinique. Et puis, qui vous dit que les biosimilaires européens sont meilleurs ? Ils ont juste moins de règles - ce qui signifie moins de sécurité. La qualité, ça ne se négocie pas.
Clementine McCrowey
Je sais que c’est dur, mais on peut faire mieux. Les patients méritent d’avoir accès à des traitements abordables. On n’a pas besoin de tout changer d’un coup - juste de réduire l’exclusivité à 8 ans. C’est réaliste. Et si on aidait les petites entreprises avec des subventions ? On peut. On doit. 💪
Jérémy allard
Les États-Unis sont une puissance. Leur système protège l’innovation. Si vous voulez des biosimilaires, allez en Europe. Mais ne venez pas nous dire que notre modèle est mauvais. On a créé ces médicaments. On les a financés. On les a développés. Et on a le droit de les protéger.
Soane Lanners
Et si tout ça était une mise en scène ? Les brevets, les biosimilaires, les 12 ans… tout est orchestré. La vérité, c’est que les grandes entreprises contrôlent les gouvernements, les agences, les médias. Les biosimilaires, c’est un piège pour vous faire croire que vous avez le choix. En réalité, vous avez juste un nouveau produit, fabriqué par un autre monopole. Le vrai problème, c’est le système capitaliste qui transforme la santé en marchandise. La vie humaine n’a pas de prix… mais dans ce monde, elle a un barème. Et il est fixé par des PDG en costume. 🕊️💸