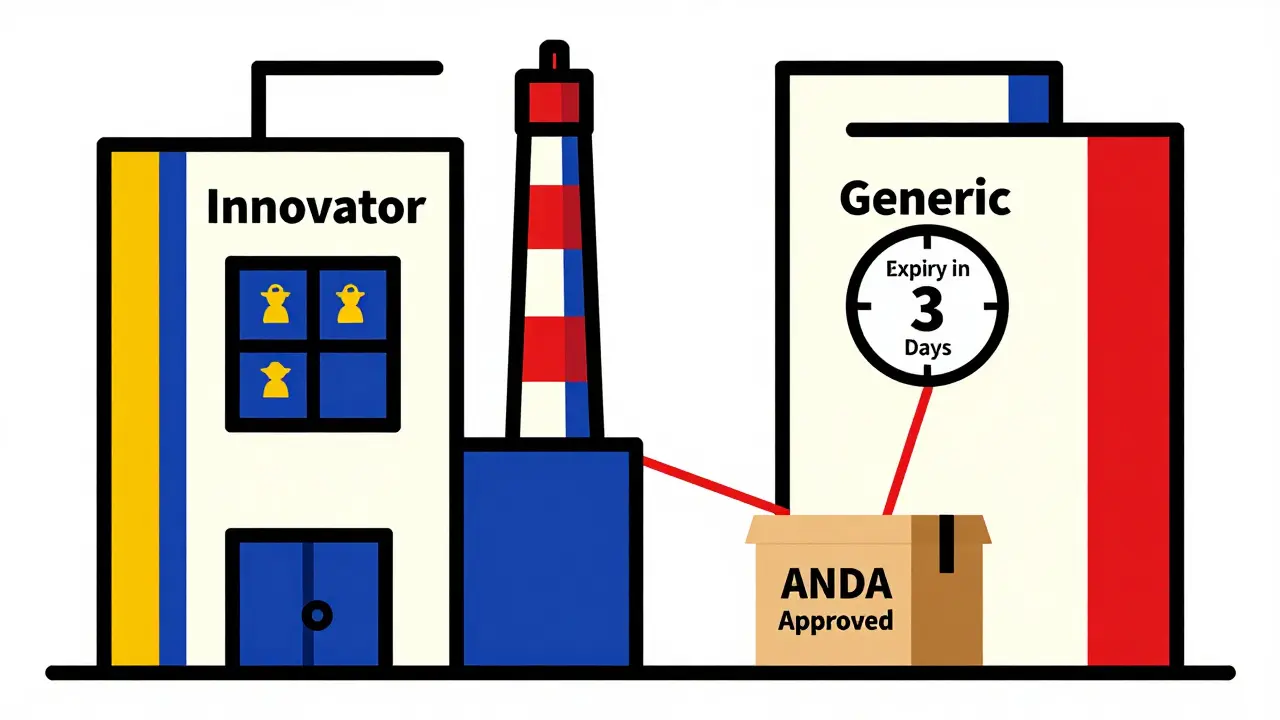
Quand une entreprise de médicaments génériques soumet une demande d’approbation à la FDA, elle ne s’attend pas à recevoir un simple « oui » ou « non ». Parfois, elle obtient un statut étrange : approbation provisoire. Ce n’est pas une autorisation de vente. Ce n’est pas non plus un refus. C’est une pause stratégique, un « vous êtes prêt, mais vous devez attendre ».
Ce système existe depuis 1984, grâce à la loi Hatch-Waxman. Elle a été conçue pour équilibrer deux mondes : celui des laboratoires innovateurs qui investissent des milliards pour créer un nouveau médicament, et celui des fabricants de génériques qui veulent proposer des versions moins chères une fois que les brevets expirent. L’approbation provisoire est le pont entre ces deux mondes. Elle permet à la FDA de valider scientifiquement un générique - ses ingrédients, sa fabrication, sa biodisponibilité - sans pour autant le laisser entrer sur le marché. Pourquoi ? Parce qu’un brevet de marque est encore en vigueur.
Imaginez que vous préparez un gâteau. Vous avez suivi la recette à la lettre, vous avez acheté les meilleurs ingrédients, vous avez testé la texture, la saveur, tout est parfait. Mais votre voisin a mis un cadenas sur la cuisine. Vous ne pouvez pas entrer. Vous n’êtes pas puni. Vous êtes juste en attente. L’approbation provisoire, c’est ça : votre gâteau est prêt. Le four est chaud. Il ne vous reste plus qu’à attendre que le cadenas tombe.
Comment ça marche ?
Le processus commence avec une demande ANDA (Abbreviated New Drug Application). Contrairement à un médicament neuf, qui demande des essais cliniques complets, un générique s’appuie sur les données du médicament original. La FDA vérifie si le générique est équivalent : même principe actif, même dose, même forme, même efficacité. Si tout est bon, mais qu’un brevet ou une exclusivité commerciale protège encore le médicament de marque, la FDA ne dit pas « non ». Elle dit : « Approbation provisoire ».
Ce n’est pas une simple lettre de félicitations. C’est un statut juridique. Le générique est « approuvé » en théorie, mais pas en pratique. Il ne peut pas être vendu aux États-Unis. Il ne peut pas être stocké en tant que produit commercial. Il est en file d’attente. Et cette file d’attente est précieuse. En 2023, la FDA a délivré environ 1 000 approbations provisoires. Chacune représente un médicament prêt à entrer sur le marché dès que la dernière barrière légale disparaît.
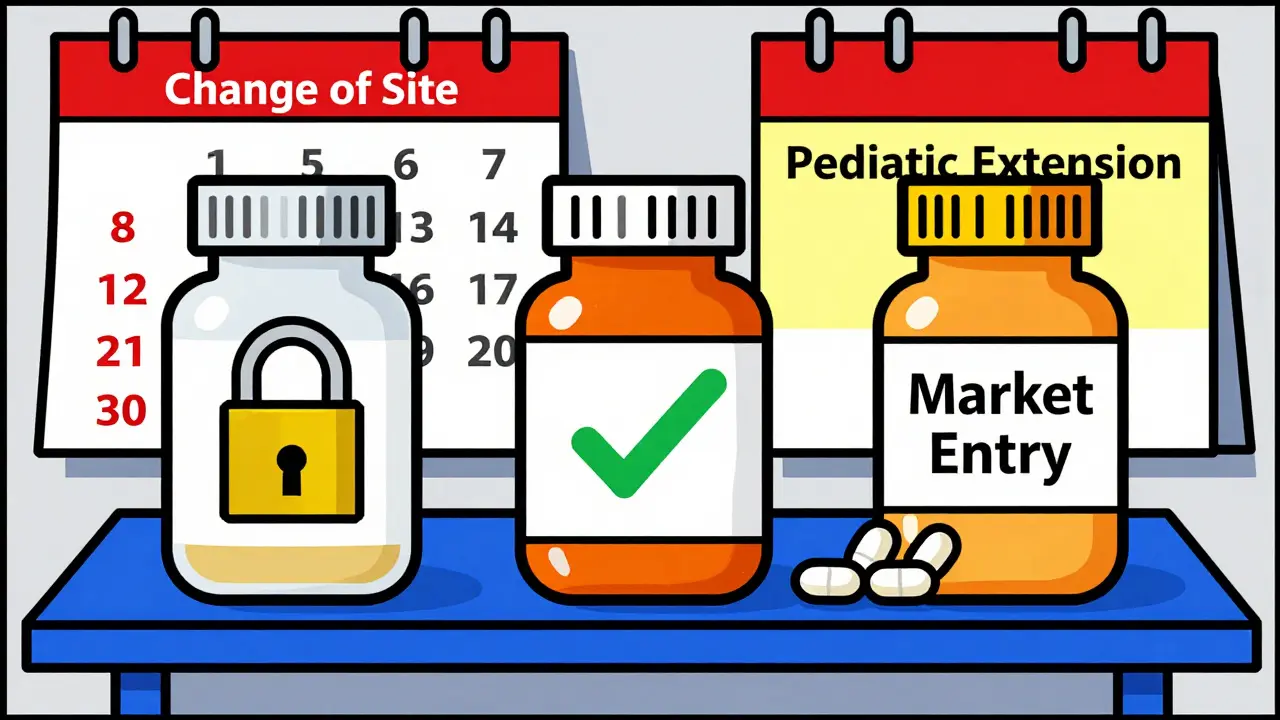
Le vrai piège, c’est de penser que tout est fini une fois qu’on a l’approbation provisoire. Ce n’est pas le cas. C’est là que les entreprises se trompent. Le processus n’est pas passif. Il exige une gestion active. La FDA exige que les entreprises soumettent des mises à jour avant la date d’expiration du brevet. Pour les changements mineurs - comme une modification de l’emballage - il faut envoyer la demande au moins trois mois à l’avance. Pour les changements majeurs - comme un nouveau site de production - il faut dix mois. Si vous oubliez, vous perdez du temps. Et du temps, c’est de l’argent perdu.
Le rôle du litige brevet
Beaucoup de génériques ne se contentent pas d’attendre. Ils attaquent. C’est ici que les certifications Paragraph IV entrent en jeu. Quand un fabricant de génériques déclare que le brevet du médicament de marque est invalide ou non applicable, il déclenche une procédure légale. Le laboratoire innovateur a 45 jours pour poursuivre en justice. Si c’est le cas, la FDA doit suspendre l’approbation finale pendant jusqu’à 30 mois - un délai appelé « stay ».
Ce n’est pas un frein. C’est un levier. Pour le premier fabricant de générique à déposer une certification Paragraph IV, la loi offre une récompense : 180 jours d’exclusivité sur le marché. Pendant ce temps, aucun autre générique ne peut entrer. C’est un jackpot. Et c’est pourquoi les entreprises investissent des millions en avocats, en experts en brevets, en analyses juridiques complexes. Certains ont réussi à faire tomber des brevets qui protégeaient des médicaments pour le VIH, le diabète, ou les maladies cardiovasculaires. D’autres ont échoué, et perdu des millions.
Le cas de Lupin Limited en 2018 est emblématique. Ils avaient obtenu l’approbation provisoire pour le générique du Cialis. Lorsque le brevet a expiré, ils ont envoyé leur demande d’approbation finale, et ont été autorisés à vendre le médicament dans les 24 heures. Ils ont capté 42 % du marché le premier mois. C’est rare, mais possible. Ce qui est plus courant, c’est l’erreur. Une entreprise a perdu six mois parce qu’elle n’avait pas compté une exclusivité pédiatrique ajoutée au brevet original. Une autre a perdu quatre mois parce qu’elle avait changé son site de production sans le signaler à la FDA. Le coût ? 150 millions de dollars de revenus manqués.

Les erreurs courantes
La plupart des entreprises pensent que l’approbation provisoire est une victoire finale. Ce n’est qu’une étape. Les erreurs les plus fréquentes sont :
- Ne pas suivre les dates d’expiration des brevets avec précision - y compris les extensions légales pour les essais pédiatriques ou les exclusivités commerciales.
- Ne pas soumettre les mises à jour à temps - surtout pour les changements de fabrication ou de composition.
- Confondre les changements mineurs et majeurs. La FDA ne fournit pas toujours des lignes claires, et les interprétations varient.
- Ne pas documenter correctement la preuve d’expiration du brevet. La FDA demande des preuves officielles, pas des estimations.
Une étude de l’Association for Accessible Medicines montre que 22 % des génériques avec approbation provisoire ont subi un retard de plus de 30 jours à l’entrée sur le marché à cause d’erreurs administratives. Ce n’est pas une question de chance. C’est une question de rigueur.
Qui utilise ce système ?
Les grands fabricants de génériques - comme Teva, Mylan, Lupin, Aurobindo - ont des équipes entières dédiées à ce processus. Ils gèrent entre 15 et 25 produits en approbation provisoire en même temps. Les petits acteurs, eux, en ont deux ou trois. Pour eux, c’est une question de survie. Un seul générique bien timed peut faire passer une entreprise de la faillite à la rentabilité.
En 2022, 85 % des génériques entrés sur le marché américain l’ont fait grâce à l’approbation provisoire. C’est la norme. Ce n’est pas un cas particulier. C’est la règle. Et cela va s’accentuer. La FDA prévoit une augmentation de 12 % par an jusqu’en 2027. Pourquoi ? Parce que les médicaments deviennent plus complexes - des biologiques, des formulations à libération prolongée, des combinaisons de plusieurs principes actifs. Ces produits ont des brevets plus longs, plus nombreux, plus difficiles à contourner. L’approbation provisoire devient encore plus essentielle.
Quel avenir ?
La FDA a récemment annoncé qu’elle réduirait le délai de traitement des demandes d’approbation finale à 30 jours pour les changements mineurs - contre 60 à 90 jours auparavant. C’est une bonne nouvelle. Mais elle ne résout pas tout. Les litiges brevets deviennent plus longs, plus techniques. Les brevets « de stratégie » - ceux qui protègent des processus de fabrication, des formes de comprimés, des combinaisons - sont de plus en plus utilisés pour repousser l’entrée des génériques.
Des projets de loi comme le « Protecting Drug Patents Act » de 2023 pourraient prolonger encore davantage les périodes d’exclusivité. Ce qui menace l’équilibre de la loi Hatch-Waxman. Pourtant, les analystes sont unanimes : l’approbation provisoire reste un pilier du système. 95 % des experts estiment qu’elle sera encore en vigueur en 2030. Parce qu’elle fonctionne. Elle permet aux patients d’avoir accès à des médicaments abordables. Elle permet aux entreprises de planifier. Elle permet à la FDA de gérer une complexité croissante sans se noyer.
Comment bien débuter ?
Si vous êtes une entreprise de génériques, voici ce qu’il faut faire :
- Soumettez une ANDA complète avec une certification Paragraph IV si vous croyez que le brevet est invalide.
- Surveillez chaque date de brevet - y compris les extensions, les exclusivités, les dérogations.
- Établissez un calendrier précis : trois mois avant l’expiration pour les changements mineurs, dix mois pour les majeurs.
- Documentez tout. Chaque changement de site, chaque modification de composition, chaque communication avec la FDA.
- Formez votre équipe. Il faut des experts en réglementation, en brevets, et en gestion de projet. La courbe d’apprentissage prend 6 à 12 mois.
Il n’y a pas de raccourci. Pas de miracle. Juste de la discipline. Et une compréhension profonde du système. Parce que dans ce jeu, le temps n’est pas seulement une ressource. C’est le seul levier qui compte.
Qu’est-ce que l’approbation provisoire de la FDA ?
L’approbation provisoire est un statut accordé par la FDA à une demande de générique (ANDA) qui répond à toutes les exigences scientifiques, mais qui ne peut pas être approuvée définitivement en raison de brevets ou d’exclusivités en cours. Cela permet à l’entreprise de préparer son entrée sur le marché dès que la barrière légale expire, sans avoir à recommencer le processus d’évaluation.
Pourquoi la FDA n’autorise-t-elle pas la vente immédiate après l’approbation provisoire ?
Parce que la loi Hatch-Waxman protège les brevets des médicaments innovants. Même si le générique est scientifiquement équivalent, il ne peut pas être commercialisé tant qu’un brevet est en vigueur. L’approbation provisoire est une reconnaissance de la qualité du produit, pas une autorisation de vente.
Quelle est la différence entre approbation provisoire et approbation finale ?
L’approbation provisoire signifie que le générique est prêt, mais qu’il ne peut pas être vendu. L’approbation finale permet la vente immédiate sur le marché américain. Le passage de l’un à l’autre ne se fait pas automatiquement : il faut soumettre une demande explicite, souvent accompagnée de mises à jour réglementaires.
Quels sont les risques si une entreprise ne gère pas bien son approbation provisoire ?
Les risques sont financiers et opérationnels : retard d’entrée sur le marché, perte de l’exclusivité de 180 jours, coût de production inutile, et même perte de la position dans la file d’attente. Des erreurs simples - comme ne pas signaler un changement de site de fabrication - peuvent entraîner des retards de plusieurs mois et des pertes de plusieurs dizaines de millions de dollars.
Comment savoir quand le brevet va expirer exactement ?
Il faut consulter les brevets déposés à l’USPTO (United States Patent and Trademark Office) et vérifier les extensions légales, comme l’exclusivité pédiatrique, les extensions de durée de brevet pour les essais cliniques, ou les protections liées à des formulations spécifiques. Aucune estimation ne suffit : seules les données officielles comptent. De nombreuses entreprises utilisent des logiciels spécialisés ou des consultants juridiques pour suivre ces dates avec précision.

8 Commentaires
marie-aurore PETIT
Je viens de finir de lire ce truc et j'ai juste envie de dire : MERCI. J'étais perdue avec toutes ces histoires de brevets et d'approbation provisoire, là je comprends enfin pourquoi certains génériques mettent des années à arriver. C'est pas juste de la bureaucratie, c'est un vrai jeu d'échecs juridique. J'adore la métaphore du gâteau et du cadenas, c'est parfait.
Mélanie Timoneda
Je trouve ça fou qu'on puisse avoir un produit 100% valide mais pas le vendre juste parce qu'un brevet est encore en vigueur. C'est comme avoir une clé pour une porte qui n'est pas encore ouverte. La loi Hatch-Waxman voulait équilibrer les choses, mais aujourd'hui, ça ressemble plus à un système où les gros jouent avec les règles. Je me demande si les patients paient vraiment moins cher à la fin.
Laetitia Ple
Alors là, je suis scotchée. 150 millions perdus parce qu'on a changé l'emballage sans prévenir la FDA ?! C'est pas une entreprise, c'est un jeu de l'oie avec des millions en jeu. Et les avocats qui se tapent des procès de 30 mois pour 180 jours d'exclusivité ? Je veux bien qu'ils soient payés à l'heure, mais bon… c'est du délire organisé.
Julien Doiron
Je ne crois pas une seule seconde que cette approbation provisoire existe pour aider les patients. Non. C'est un système conçu par les lobbys pharmaceutiques pour ralentir délibérément l'accès aux génériques. Les brevets pédiatriques ? Des inventions juridiques pour prolonger les monopoles. La FDA est complice. Tout cela est orchestré pour que les prix restent élevés. Les gens meurent parce qu'ils ne peuvent pas acheter leurs médicaments. Et vous, vous parlez de « processus » comme si c'était une question de paperasse. C'est un crime.
Louis Ferdinand
Le truc qui m'a frappé, c'est que 85 % des génériques entrent sur le marché via cette approbation provisoire. Donc c'est pas un cas spécial. C'est le standard. Et pourtant, personne n'en parle. On parle des médicaments, pas de leur parcours juridique. C'est fou qu'une telle machine existe, invisible, et qu'elle permette à des milliers de gens d'avoir accès à des traitements abordables. On oublie trop souvent que la santé, c'est aussi de la logistique.
Laurence TEIL
En France, on n'a pas ce genre de dérives. Ici, les génériques sont contrôlés, les prix sont fixés, et on n'attend pas des années pour que les brevets expirent. Ce système américain est une aberration. On laisse des entreprises se battre comme des chien pour un droit de vente ? On est dans le capitalisme sauvage. Et vous, vous trouvez ça normal ? Je vous le demande sérieusement. En France, on protège les patients, pas les avocats.
Mats During
Vous savez ce qui est vraiment incroyable ? Ce système n'existe que parce que les États-Unis sont le seul marché où les brevets peuvent être contestés par des entreprises privées. En Europe, on a des procédures administratives, pas des guerres juridiques. Ici, c'est un champ de bataille. Les entreprises ne cherchent pas à innover, elles cherchent à trouver des failles dans les brevets. Le vrai génie, ce n'est pas de créer un médicament, c'est de trouver un mot dans un brevet qui n'a pas été écrit correctement. Et ça, c'est la déchéance de l'innovation. On a remplacé la science par la syntaxe. C'est triste. Très triste.
Sabine Schrader
Wow. Juste… wow. Merci pour ce texte incroyablement clair !!!! C'est tellement important de comprendre ça, surtout quand on a des proches qui dépendent de ces médicaments!!! J'ai appris tellement de choses!!! Le fait que la FDA exige des mises à jour trois mois avant… c'est fou comment une petite erreur peut tout casser!!! Je vais partager ça à tout le monde!!!